
Breathing New Life into Idiopathic Pulmonary Fibrosis: Advances in Understanding a Complex Lung Disease

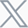
Idiopathic pulmonary fibrosis (IPF) is an intriguing yet perplexing lung disease that has captured the attention of scientists and medical professionals alike. Characterized by inflammation and scarring of the delicate air sacs in the lungs, known as alveoli, IPF is a debilitating chronic condition. The scarring process, called fibrosis, leads to the stiffening of lung tissue, impairing its ability to efficiently absorb oxygen from the air. As a result, individuals with IPF face difficulties in breathing, greatly impacting their quality of life.
A comprehensive systematic review and meta-analysis revealed that the global occurrence of IPF ranges from 0.09 to 1.30 cases per 10,000 individuals[1]. Additionally, the review highlighted the age-standardized mortality rate of IPF, which varies from 0.5 to 12 cases per 100,000 individuals. It is concerning to note that the mortality rate of IPF has been increasing in many countries over time. These findings underscore the urgent need for enhanced understanding, research, and effective interventions to address the growing burden of IPF on individuals and societies worldwide.
Age- and sex-stratified prevalence of idiopathic pulmonary fibrosis[2]
While the precise cause of this disease remains unknown, certain factors have been associated with its development. Aging is one such factor, as IPF tends to affect individuals over the age of 50. Environmental exposures, such as occupational hazards or a history of cigarette smoking, have also been implicated. Interestingly, genetics may play a role, as some cases of IPF seem to run in families, pointing to possible genetic predispositions.
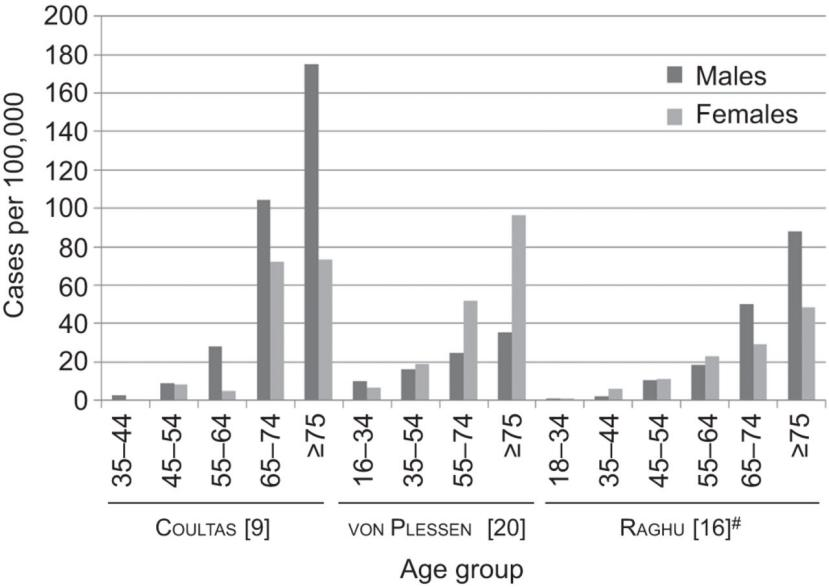
To comprehend IPF better, we must delve into the intricate workings of the lungs. The pathological mechanism underlying IPF is a complex and dynamic process involving various cell types and molecular pathways. Although not fully understood, it revolves around an intricate interplay of cells and signaling pathways that result in repetitive injury to alveolar epithelial cells (AECs) and subsequent activation of fibroblasts and myofibroblasts. These activated cells produce an excessive amount of extracellular matrix (ECM), leading to fibrosis and stiffening of lung tissue, ultimately impairing gas exchange and breathing. Let's delve deeper into the stages of this pathological process[3,4].
Healthy lungs exhibit intact alveolar basement membranes that facilitate efficient gas exchange, ensuring effective oxygen uptake and carbon dioxide release. However, when exposed to environmental triggers or genetic predispositions, AECs can undergo apoptosis, necrosis, or senescence. Injured AECs release profibrotic factors, including transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), and fibroblast growth factor (FGF). These factors stimulate the activation and migration of fibroblasts and myofibroblasts to the injury site. Subsequently, activated fibroblasts and myofibroblasts synthesize and deposit ECM components, such as collagen, elastin, and fibronectin, within the lung interstitium. This abnormal ECM accumulation disrupts alveolar architecture and reduces lung tissue compliance. Over time, the accumulated ECM forms fibrotic foci, dense areas of fibrosis in the lung interstitium. These foci distort alveolar structure, impairing gas exchange. With progression, the fibrotic foci may coalesce, forming honeycomb cysts, which are characteristic features of advanced or end-stage idiopathic pulmonary fibrosis (IPF). These cysts, characterized by thickened walls and dilated airspaces, further contribute to the loss of lung function.
Schematic summary of IPF pathogenesis[5]
When it comes to battling IPF, a comprehensive and multidisciplinary approach is necessary. Early diagnosis is crucial in preventing further damage. High-resolution computed tomography (HRCT) scans and pulmonary function tests are commonly used to evaluate lung function and assess the extent of fibrosis. These tests help tailor treatment plans to individual patients. While a cure for IPF remains elusive, several treatment options aim to slow its progression and effectively manage symptoms. Pulmonary rehabilitation programs take a holistic approach by combining exercise, breathing techniques, and education to improve quality of life. Oxygen therapy can alleviate breathlessness and improve oxygen levels. In more advanced cases, lung transplantation can be considered for eligible patients.
In recent years, significant progress has been made in the pharmacological management of IPF. Antifibrotic medications, such as pirfenidone and nintedanib, have shown promising results in clinical trials. Nintedanib, a potent kinase inhibitor, blocks the effects of growth factors involved in IPF pathogenesis, such as PDGF, FGF, and VEGF. Pirfenidone, an antifibrotic medication, targets the underlying fibrotic processes in IPF by reducing the production of pro-fibrotic factors like TGF and inhibiting ECM synthesis. Clinical trials have demonstrated that both medications can slow the progression of IPF over a 52-week period, as measured by changes in forced vital capacity (FVC)[6,7].
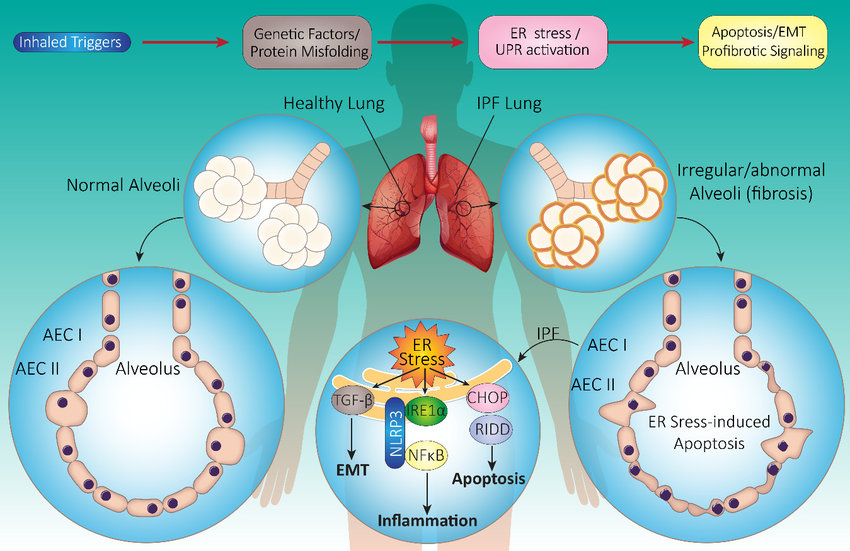
Despite these advancements, IPF remains a puzzling enigma for researchers worldwide. Ongoing scientific investigations aim to unravel the complex mechanisms underlying the disease, searching for new therapeutic approaches and personalized treatment strategies. Disease models play a crucial role in scientific exploration. GemPharmatech utilizes a non-invasive inhalation method to deliver a pharyngeal aspirate of a BLM-containing PBS solution into the lungs of mice. This approach significantly reduces mouse mortality compared to traditional intubation or tracheal instillation methods, enabling efficient model construction.
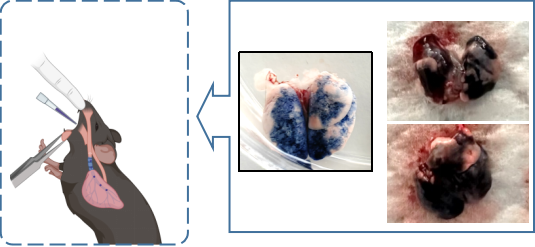
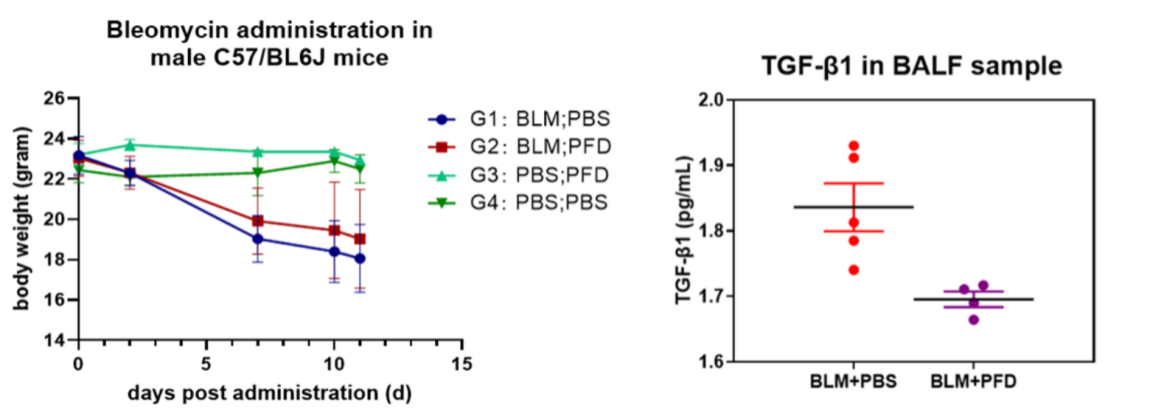
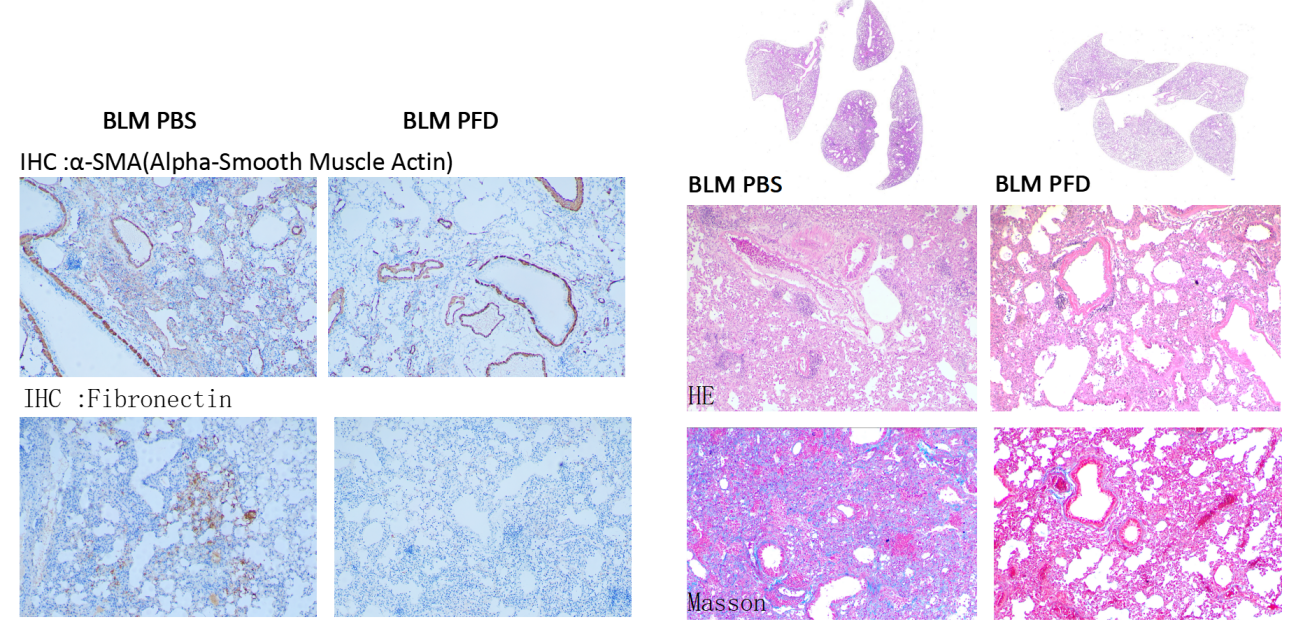
Following PFD treatment, no evident toxic effects were observed, as indicated by body weight. ELISA analysis of mouse lung lavage fluid revealed reduced levels of TGF-β, suggesting a mitigating effect on pulmonary fibrosis. Immunohistochemistry analysis demonstrated a significant decrease in the expression of α-SMA (fibroblast marker, stained brown) and fibronectin (fibrosis marker, stained brown). Histological staining using HE and Masson's trichrome staining exhibited noticeable improvement in interstitial pneumonia phenotypes, including reduced neutrophil infiltration foci and pulmonary consolidation, as well as attenuation of BLM-induced fibrosis.
Contact us today to learn more about our IPF models!
Reference:
1. Zheng Q, Cox IA, Campbell JA, Xia Q, Otahal P, de Graaff B, Corte TJ, Teoh AKY, Walters EH, Palmer AJ. Mortality and survival in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. ERJ Open Res. 2022 Mar 14;8(1):00591-2021.
2. Barratt SL, Creamer A, Hayton C. Idiopathic pulmonary fibrosis: Global incidence and prevalence. BMJ Open Respir Res. 2015;2(1):e000005.
3. Moss BJ, Ryter SW, Rosas IO. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu Rev Pathol. 2022 Jan 24;17:515-546.
4. Zhang Y, Li Y, Liu Y, et al. Recent advances on the pathogenesis of idiopathic pulmonary fibrosis and novel therapeutic strategies. Front Pharmacol. 2022;12:814.
5. Aghaei, Mahmoud D, Sanaz A, et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life. 2020 Nov 10.3390/life11010001.
6. Marijic P, Schwarzkopf L, Schwettmann L, Ruhnke T, Trudzinski F, Kreuter M. Pirfenidone vs. nintedanib in patients with idiopathic pulmonary fibrosis: a retrospective cohort study. Respir Res. 2021 Oct 19;22(1):268.
7. Finnerty, J.P., Ponnuswamy, A., Dutta, P. et al. Efficacy of antifibrotic drugs, nintedanib and pirfenidone, in treatment of progressive pulmonary fibrosis in both idiopathic pulmonary fibrosis (IPF) and non-IPF: a systematic review and meta-analysis. BMC Pulm Med 21, 411 (2021).