
Breaking Barriers: Advancing Gene Therapies for Amyotrophic Lateral Sclerosis

Amyotrophic Lateral Sclerosis (ALS) is a relentless and complex neurodegenerative disorder that affects the central nervous system, gradually impairing voluntary muscle movement. Referred to as Lou Gehrig's disease, ALS has fascinated the scientific and medical communities due to its perplexing nature.
ALS is characterized by the degeneration of motor neurons responsible for transmitting signals from the brain to the muscles throughout the body. This progressive loss of motor neurons leads to muscle weakness, paralysis, and eventually respiratory failure. Patients experience a range of symptoms, including muscle cramps, muscle twitching (fasciculations), difficulty swallowing (dysphagia), slurred speech (dysarthria), and muscle stiffness (spasticity). The relentless progression of ALS presents immense challenges for those affected, necessitating a deeper understanding of the disease.
ALS is a global health concern, affecting people of all ethnicities and geographic regions. According to a systematic review and meta-analysis[1], the overall crude worldwide incidence of ALS was 1.59 per 100,000 person-years. The prevalence of ALS increases with age, with the majority of cases diagnosed between the ages of 40 and 70.
Currently, ALS treatment therapies primarily focus on managing symptoms and improving patients' quality of life. There are three approved medications: riluzole, edaravone, and Relyvrio, two of which have shown moderate efficacy, resulting in a slight increase in survival of several months[2, 3]. Additionally, supportive therapies such as physical and occupational therapy, speech therapy, and assistive technologies help maintain functionality and autonomy. It is important to note, however, that these therapeutic options do not halt or reverse the progression of the disease. This underscores the critical importance of exploring novel techniques to address ALS's relentless progression in the fight against the disease.
While the majority of ALS cases develop spontaneously, approximately 5-10% of cases exhibit familial patterns, suggesting an underlying genetic predisposition. The identification of SOD1 (superoxide dismutase 1) as the first causal gene for ALS dates back to 1993[4]. Since then, a number of genes, including C9orf72, TARDBP, FUS, and others, have been linked to the pathophysiology of ALS. These genes play crucial roles in protein regulation, RNA metabolism, and cytoskeleton organization, among other cellular functions. By elucidating the genetic basis of ALS, we not only gain a better understanding of the disease but also open up new avenues for targeted therapeutics and personalized medicine, paving the way for more precise interventions.
In order to gain a better understanding of ALS pathogenesis and develop effective treatments, mouse models play a crucial role. GemPharmatech has developed a proprietary mouse model named B6-hSOD1 G93A, which carries the full-length human SOD1 gene with the G93A mutation, including the human promoter sequence, exons, and introns. This model proves invaluable in evaluating ASO drugs targeting the SOD1 pathway and other ALS therapies. Preliminary validation results demonstrate that this mouse model partially replicates the clinical and behavioral characteristics observed in ALS patients, with disease onset occurring at approximately 5.5 months of age. As a result, it serves as an ideal research tool for investigating ALS pharmacology, drug efficacy, and disease mechanisms.
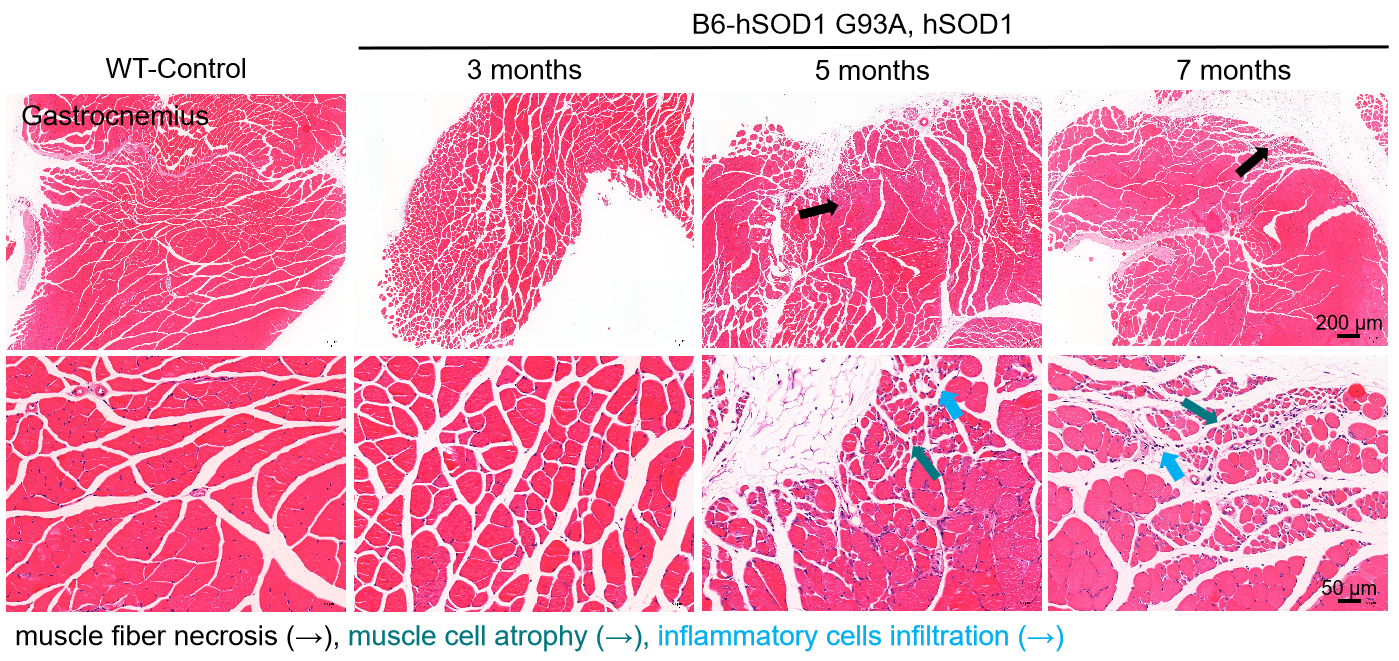
Figure 1. Muscle histopathology of B6-hSOD1 G93A, hSOD1 male mice
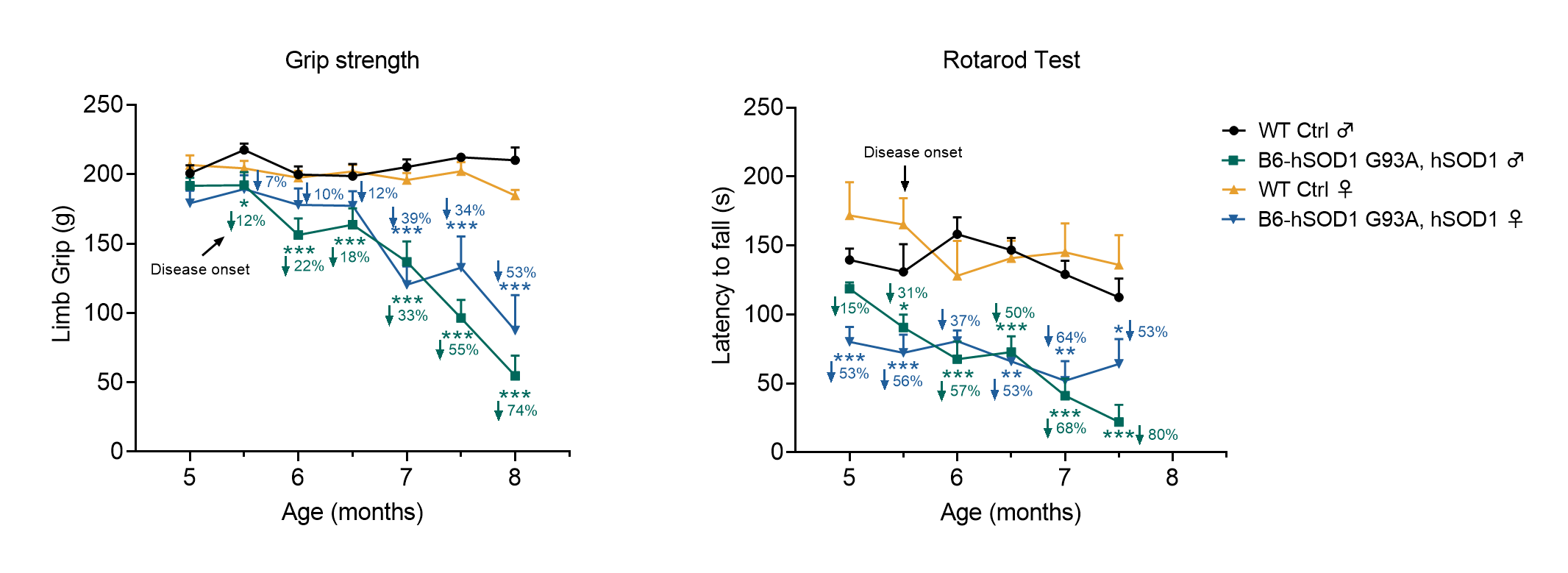
Figure 2. Motor capability changes in B6-hSOD1 G93A, hSOD1 mice
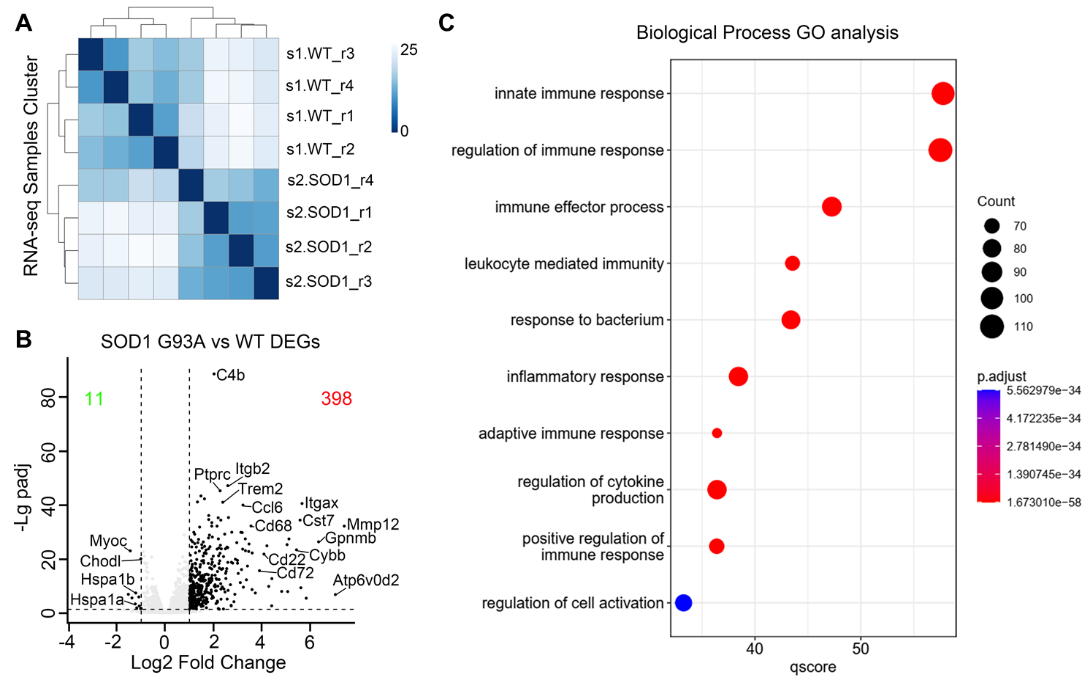
Figure 3. Transcriptome expression profile changes in the spinal cord of B6-hSOD1 G93A, hSOD1 mice at the disease onset
Aside from the well-studied SOD1 gene, GemPharmatech has successfully developed a proprietary mouse model, known as B6-hTDP43 A315T, which specifically targets another crucial gene associated with ALS, TARDBP. This model proves to be an invaluable resource for comprehending the involvement of the TARDBP gene or TDP43 protein in ALS, as well as for evaluating drug effectiveness and elucidating disease mechanisms.
References:
1. Xu L, Liu T, Liu L, Yao X, Chen L, Fan D, Zhan S, Wang S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol. 2020 Apr;267(4):944-953.
2. Jaiswal, M.K., Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev, 2019. 39(2): p. 733-748.
3. Chio, A., L. Mazzini, and G. Mora, Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology, 2020. 167: p. 107986.
4. Deng, H.X., Hentati, A., Tainer, J.A., et al. (1993). Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261, 1047–1051.